

Vědci objevili biologický mechanismus i účinnou látku na léčbu dědičného chronického onemocnění ledvin – ADTKD. Látka musí projít ještě klinickými testy, ale již nyní se jeví jako účinná pro celou skupinu nemocí zvaných proteinopatie. O objevu informoval prestižní americký časopis Cell.
 Předběžné výsledky výzkumu dědičně podmíněných onemocnění ledvin jsou zlomové. Pokud je potvrdí klinické testy, bude to mimo jiné znamenat, že pacienti se vzácným onemocněním ADTKD nebudou muset spolu s ostatními čekat na transplantaci ledvin. Předběžné výsledky výzkumu prokázaly, že vědci objevená látka, může pomoci i pacientům s dědičným poškozením sítnice oka. Další dobrá zpráva je, že v týmu, který přišel se senzačním objevem, působili i čeští lékaři z 1. lékařské fakulty Univerzity Karlovy.
Předběžné výsledky výzkumu dědičně podmíněných onemocnění ledvin jsou zlomové. Pokud je potvrdí klinické testy, bude to mimo jiné znamenat, že pacienti se vzácným onemocněním ADTKD nebudou muset spolu s ostatními čekat na transplantaci ledvin. Předběžné výsledky výzkumu prokázaly, že vědci objevená látka, může pomoci i pacientům s dědičným poškozením sítnice oka. Další dobrá zpráva je, že v týmu, který přišel se senzačním objevem, působili i čeští lékaři z 1. lékařské fakulty Univerzity Karlovy.
Jak to začalo
Před 20 lety vyšetřovali lékaři českou rodinu. Její členové měli zvýšené hladiny kyseliny močové v krvi, trpěli dnou [zánětlivé postižení kloubů] a progresivním selháním ledvin. To vedlo k dialýze a transplantaci ledvin. Na konci 90. let bylo známo ve světě několik desítek rodin s touto nemocí. Onemocnění bylo dominantně dědičné [50% pravděpodobnost]. Klinické projevy onemocnění u tehdy zkoumané rodiny vedly ke vzniku různých názvů nemoci – familiární hyperurikemická nefropatie nebo medulární cystické onemocnění ledvin. Lékaři nevěděli, co je příčinou tohoto onemocnění, ale později přišli na to, že se jedná o genovou mutaci.
„V té době jsme u nás na pracovišti začali používat metody a postupy takzvaného pozičního klonování, které nám umožňovaly odhalit genetické příčiny širokého spektra dědičně podmíněných onemocnění. Pomocí těchto postupů se nám postupně ve zmíněné a dalších rodinách podařilo definovat čtyři geny UMOD, REN, MUC1 a SEC61A1, jejichž mutace tento typ onemocnění ledvin způsobují,“ vzpomíná na začátky výzkumu klíčová členka řešitelského týmu Martina Živná.
Podle ní znalost genetických příčin nemoci a možnost jejich genetické diagnostiky vedly k zavedení nového světově uznávaného názvu nemoci. To znamená autozomálně dominantní tubulointersticiální nefropatie [ADTKD] s přívlastkem příslušné genetické příčiny [ADTKD-UMOD, ADTKD-REN, ADTKD-MUC1 a ADTKD-SEC61A1]. Podle Martiny Živné pojmenování nemoci bylo prvním krokem k jejímu dalšímu zkoumání.
„Spolu s profesorem Anthonym J. Bleyerem z Wake Forest School of Medicine ve Spojených státech, který je zároveň hostujícím profesorem 1. LF UK, a dále s kolegy z Broad Institute of MIT and Harvard, v Cambridge a z University of Cyprus jsme vytvořili velmi efektivní pracovní skupinu, která se začala problematice ADTKD věnovat,“ dodává.
Jak to pokračovalo
Anthony J. Bleyer byl tím, kdo zajišťoval klinickou část výzkumu. Systematicky shromažďoval rodiny i pacienty z celého světa. To výzkumnému týmu umožnilo systematické hledání charakteristiky genetické příčiny onemocnění ADTKD a příbuzných nemocí.
„Postupně jsme po celém světě diagnostikovali a charakterizovali ADTKD u cca 665 rodin. Což činí z ADTKD druhé nejpočetnější genetické onemocnění ledvin po polycystóze,“ říká Anthony J. Bleyer.
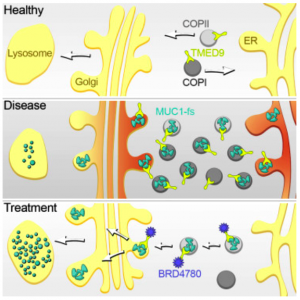
Vedoucí týmu vědců za 1. LF UK byl Stanislav Kmoch. Podle něj byla primárním cílem výzkumu nemoc ADTKD-MUC1. Ta je způsobena specifickým typem „posunových“ mutací v genu MUC1. Ty vedou ke vzniku abnormálního, tělu cizímu, proteinu MUC1-fs v buňkách ledvinných kanálků. Postižené buňky si neumí s MUC1-fs poradit. Ten se v nich střádá a postupně buňky a ledviny ničí.
„Hlavním cílem výzkumu bylo tedy objasnit, kde a proč se MUC1-fs v buňkách střádá, jaké biologické dopady má střádání na buňky, ledvinu a organismus a nalezení látky či látek, které by zamezily produkci MUC1-fs nebo umožnily jeho odbourávání,“ vysvětluje.
Zjednodušeně řečeno byl výzkum rozdělen do tří fází. První spočívala v odhalení genetických příčin onemocnění, druhá v objasnění molekulárních mechanismů vzniku onemocnění, třetí pak v hledání léčby, která by rozvoj selhání ledvin oddálila, optimálně úplně zastavila.
Hledání léčebné látky
Tým, jež se v rámci mezinárodní multioborové spolupráce zabýval objevením látky, která by pacientům mohla zachránit ledviny, vedla Anna Greka z Broad Institutu. Účinnou látku spolu s kolegy vyvíjela sedm let.
„Díky tomu, že jsme byli v aktivním kontaktu s pacienty po celém světě, jsme mohli zkoumat MUC1-fs v bioptických materiálech, imortalizovaných buněčných kulturách ledvin a v ledvinných organoidech připravených z indukovaných pluripotentních buněk pacientů,“ vysvětluje.
Upřesňuje, že k výzkumu využili i geneticky modifikované myši, které produkují lidský MUC1-fs a měli projevy postižení ledvin.
„Studium výsledků testů vedlo k poznatku, že se MUC1-fs v buňce střádá hlavně v TMED9 pozitivních transportních váčcích. TMED9 je protein, který kontroluje kvalitu vnitrobuněčného transportu proteinů,“ upřesňuje.
Buněčné modely byly dle jejich slov následně podrobeny účinku 3 500 látek, které byly již v minulosti testovány za různým účelem v různých fázích klinických studií na lidech. Jednalo se o ideální látky, protože měly potenciál přeskočit několik fází klinických studií a dostat se v kratším čase do klinické praxe.
Objev BRD4780
„Screening přitom odhalil jednu látku [BRD4780, pozn. red.], která jak buňkám, tak i geneticky modifikovaným myším umožnila MUC1-fs, jež způsobuje onemocnění, velmi efektivně odstraňovat,“ vysvětluje Anna Greka.
To, jak BRD4780 funguje ve vztahu k MUC1-fs, doplňuje Stanislav Kmoch: „BRD4780 se přímo váže na TMED9. Tím snižuje jeho kontrolní kapacitu a umožňuje buňce posunout MUC1-fs z transportních váčků do lysozomu. Tedy místa, kde buňky degradují nepotřebné proteiny.“
Logickou otázkou pak podle něj bylo, zda se mechanismus a efekt BRD4780 uplatňuje i u dalších lidských nemocí spojených s poruchou transportu jiných mutovaných proteinů, tzv. toxických proteinopatií.
„Předběžné studie prokázaly stejný mechanismus a efekt u ADTKD-UMOD a také u dědičného poškození sítnice oka,“ uzavírá s tím, že prozatím nelze odhadnout, zda a kdy se nově objevená látka dostane do klinického testování.
Co je ADTKD
ADTKD je dědičné chronické onemocnění ledvin. Projevuje se postupnou ztrátou jejich funkce. Ztráta funkce nepostihuje množství produkované moče, ale filtrační schopnost ledvin. Pacienti s daným onemocněním potřebují dialýzu nebo transplantaci ve věku 30 až 60 let.
Onemocnění způsobuje mutace v jednom z genů, které mají vztah ke správné funkci ledvin. Jedná se buď o gen zvaný UMOD, REN, MUC1 nebo SEC61A1. Každý z těchto genů je předlohou pro vytvoření proteinu, jehož správná funkce se podílí na správné funkci ledvin. Nejčastěji je onemocnění podmíněno mutací buď v MUC1 nebo UMOD genu, což následně vede ke změně buď v proteinu mucin 1 [MUC1] nebo uromodulinu [UMOD]. Méně často v reninu [REN] nebo SEC61A.
První příznakem chronického dědičného onemocnění ledvin je vzrůstající kreatinin. V některých případech onemocnění ledvin může být mírný vzrůst hodnoty kreatininu naměřen už v dětství. Hodnota kreatininu v krvi pacientů s přibývajícím věkem pomalu vzrůstá. Rychlost růstu kreatininu se může u každého pacienta lišit.
Dnové záchvaty
Z důvodu vzrůstajícího množství kyseliny močové v krvi [urikémie] se může kyselina močová začít ukládat v kloubech a způsobovat dnu. Pro dnu jsou typické záněty kloubů především palců u nohou, kotníků, kolenou a loktů.
Dnavý záchvat je velmi bolestivý a zasažené klouby jsou horké [zapálené]. Ne každý s dědičným chronickým onemocněním ledvin má dnu [hyperurikémii], ale je to jeden z velmi častých příznaků. Přestože dna je poměrně časté onemocnění, tak brzký věk nástupu je neobvyklý, varující, neboť obvykle se dna objevuje ve středním až pozdějším věku [kolem 40 let].















{kind=link}